This article walks through the standard post-inference analysis workflow: annotated tree → diagnostic PR curve → confidence trimming → clone assignment → visualization.
Core concepts for interpretation
- Initial tree topology: a point-estimate starting tree constructed using neighbor joining (NJ) on continuous VAF matrices. This provides a fully-resolved (binary) initialization that empirically captures strong lineage signal before posterior sampling.
-
Posterior clade support: per-node support values in
tree$node.label(0–1) estimated from MCMC topology sampling. -
Confidence-based topology refinement: collapse
internal edges below a support cutoff
τto obtain a refined lineage tree.
Setup
We use a small in vitro LARRY barcode sample (200 cells, 186 variants) bundled with the package.
Run the same inference command used in the README:
Rscript inst/bin/run_mitodrift_em.R \
--mut_dat inst/extdata/pL1000_mut_dat.csv \
--outdir mitodrift_demo \
--tree_mcmc_iter 5000 \
--tree_mcmc_chains 4 \
--tree_mcmc_burnin 1000This writes mitodrift_demo/mitodrift_object.rds and
mitodrift_demo/tree_annotated.newick. The workflow below
uses the same mutation table and an annotated tree generated with those
settings.
mut_dat <- read.csv(
system.file("extdata", "pL1000_mut_dat.csv", package = "mitodrift")
)
data(pL1000_tree_annot)
tree_annot <- pL1000_tree_annotVisualize the full binary tree
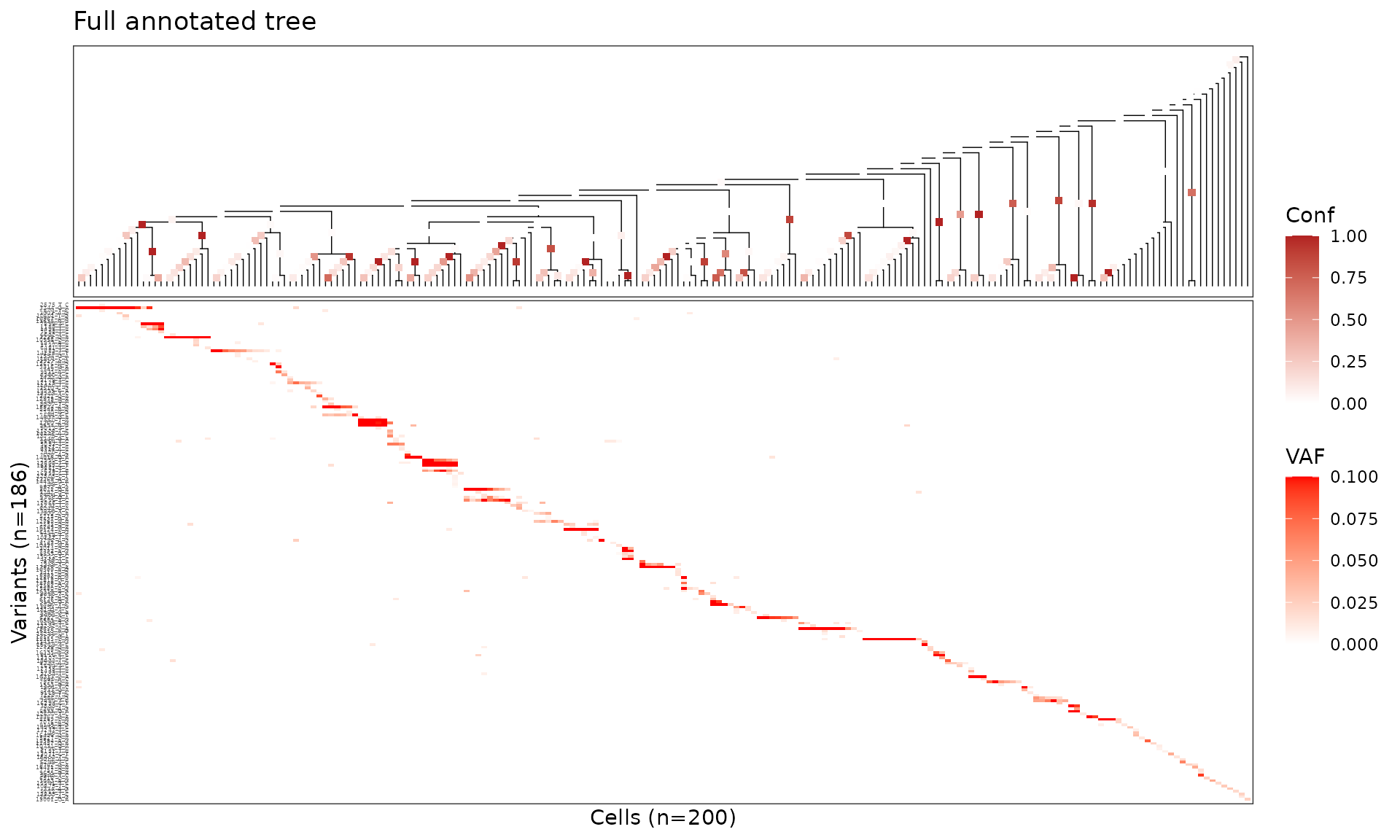
plot_phylo_heatmap2() displays the tree alongside a
variant heteroplasmy heatmap. Setting node_conf = TRUE
colours internal nodes by their confidence score.
plot_phylo_heatmap2(
tree_annot,
mut_dat,
node_conf = TRUE,
dot_size = 2,
branch_length = FALSE,
title = "Full annotated tree"
)
Diagnostic: variant precision–recall curve
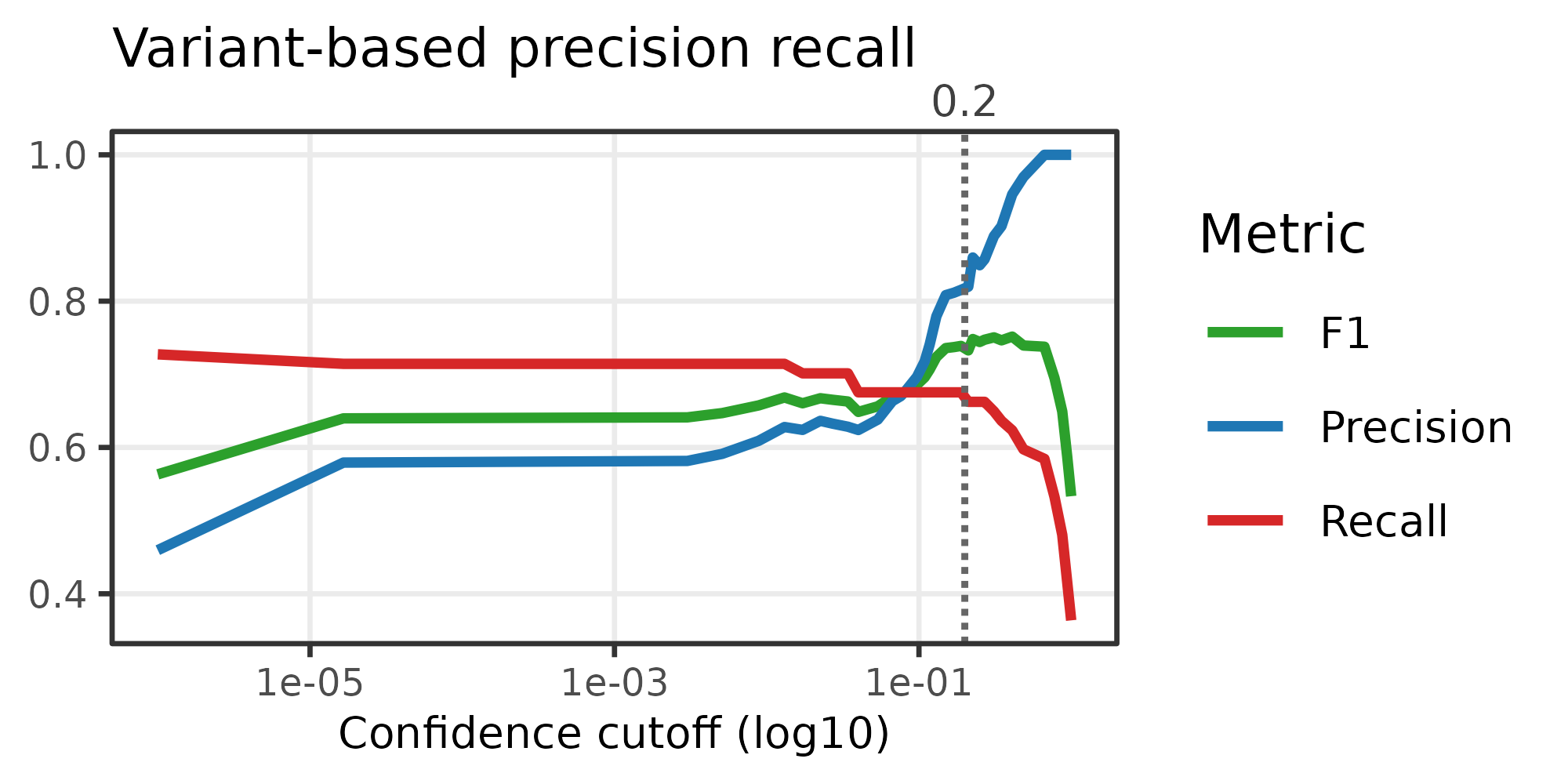
compute_variant_pr_curve() compares variant-defined cell
partitions against tree clades across a sweep of confidence cutoffs.
This helps identify a threshold that balances precision (are the clades
real?) and recall (are we keeping enough structure?).
pr_df <- compute_variant_pr_curve(tree_annot, mut_dat)
plot_prec_recall_vs_conf(
pr_df,
sample_name = "Variant-based precision recall",
cutoff = 0.2
)
Trim tree
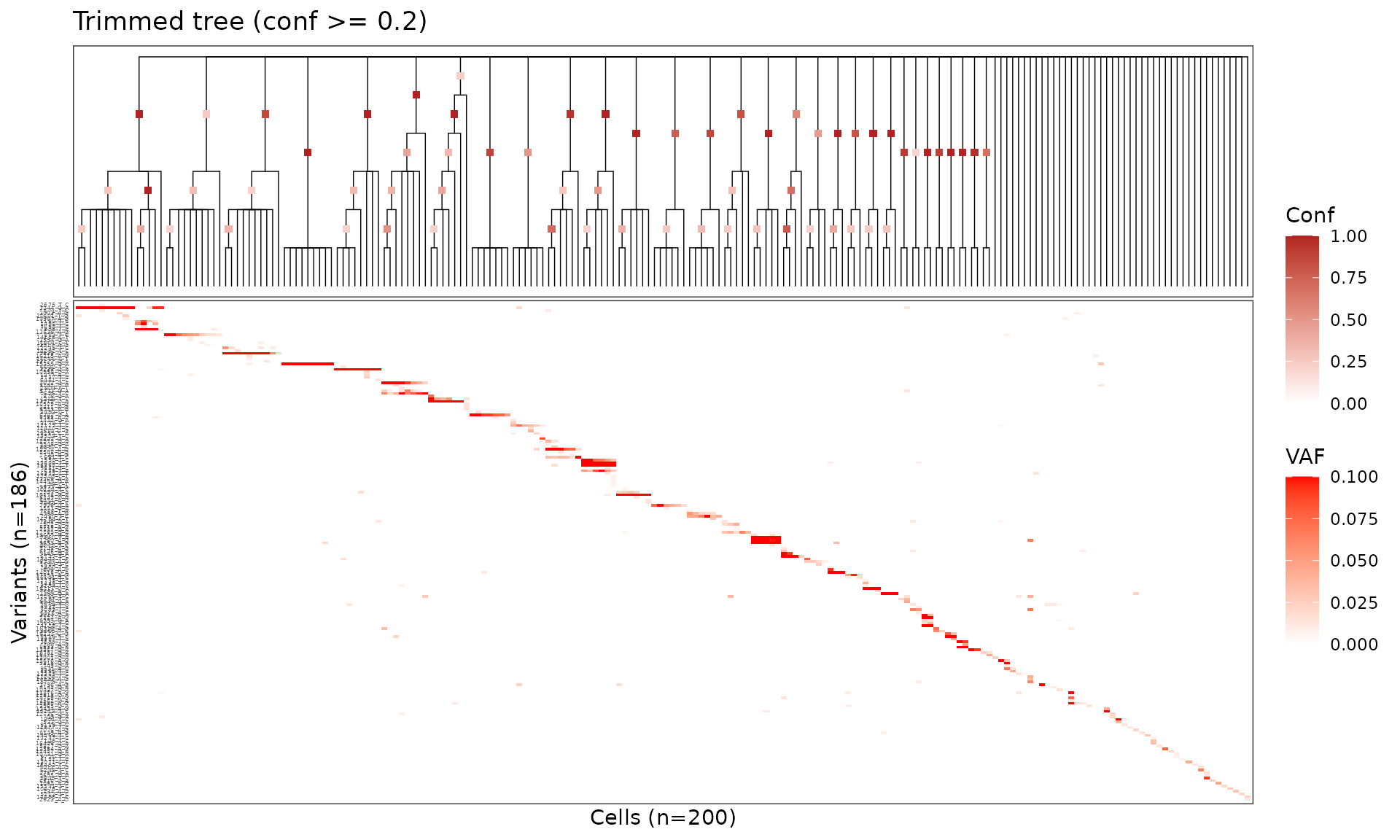
Collapse low-confidence nodes below the chosen threshold with
trim_tree().
tree_trim <- trim_tree(tree_annot, conf = 0.2)Visualize the trimmed tree — polytomies replace poorly supported splits.
plot_phylo_heatmap2(
tree_trim,
mut_dat,
node_conf = TRUE,
dot_size = 2,
branch_length = FALSE,
title = "Trimmed tree (conf >= 0.2)"
)
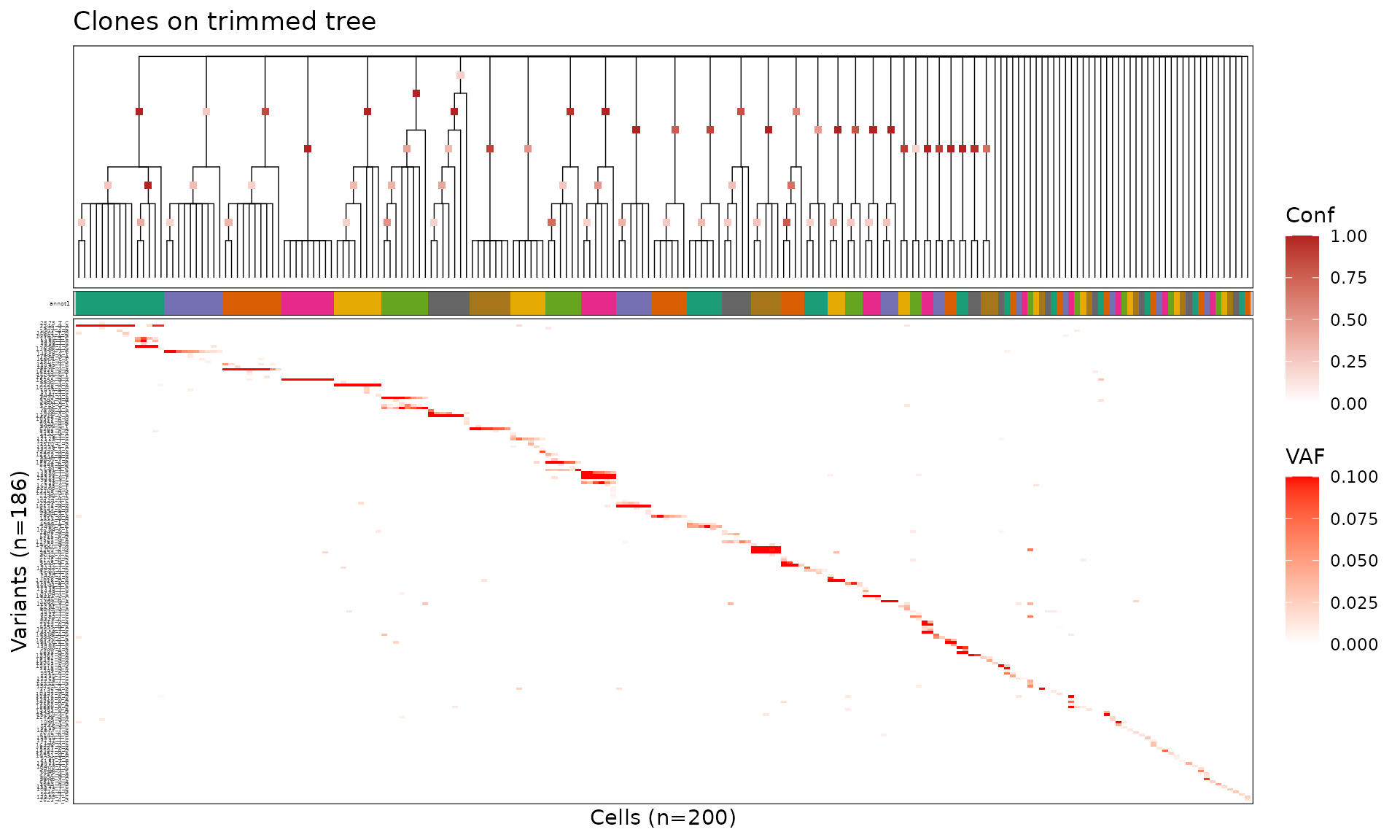
Clone assignment
assign_clones_polytomy() partitions tips into clones
based on the polytomy structure of the trimmed tree.
clone_df <- assign_clones_polytomy(tree_trim)
head(clone_df)## # A tibble: 6 × 6
## cell clade clade_node annot size frac
## <chr> <chr> <int> <chr> <int> <dbl>
## 1 CAACTAATCATTGACA-1 1 202 1 15 0.075
## 2 GTTCATTTCGGTTTGG-1 1 202 1 15 0.075
## 3 ATGTAAGCAATTGCGC-1 1 202 1 15 0.075
## 4 GGGCAATAGGCCCAGT-1 1 202 1 15 0.075
## 5 CAGCCTAAGACAACAG-1 1 202 1 15 0.075
## 6 TAGGCTAGTCGAAGTC-1 1 202 1 15 0.075Visualize clones
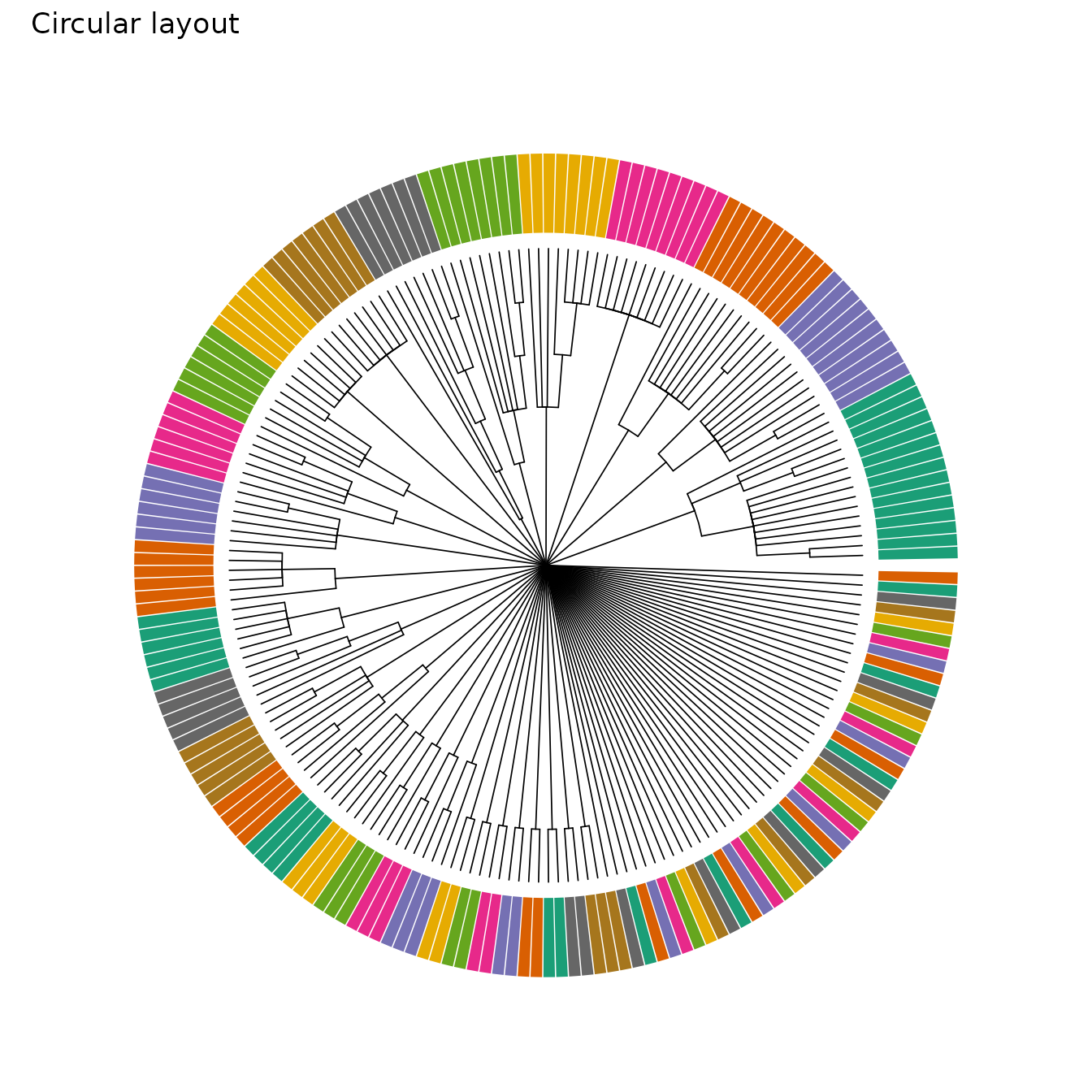
Colour cells by clone assignment on both a rectangular heatmap view and a circular layout.
clade_order <- unique(clone_df$clade)
clone_pal <- make_clade_pal(length(clade_order), labels = clade_order,
pal = "Dark2", cycle_len = 8, cycle_shift = 0)
plot_phylo_heatmap2(
tree_trim,
mut_dat,
cell_annot = clone_df,
annot_pal = clone_pal,
node_conf = TRUE,
dot_size = 2,
branch_length = FALSE,
title = "Clones on trimmed tree"
)
plot_phylo_circ(
tree_trim,
cell_annot = clone_df,
annot_pal = clone_pal,
annot_legend = FALSE,
title = "Circular layout"
)